Medical term:
mucoviscidosis
cystic
[sis´tik]1. pertaining to or containing cysts.
2. pertaining to the urinary bladder or to the gallbladder.
cystic disease of breast fibrocystic disease of breast.
cystic fibrosis a hereditary disorder associated with widespread dysfunction of the exocrine glands, with accumulation of excessively thick and tenacious mucus and abnormal secretion of sweat and saliva; it is inherited as a recessive trait; both parents must be carriers. The cause is thought to be absence, insufficiency, or abnormality of some essential hormone or enzyme. Called also cystic fibrosis of the pancreas and mucoviscidosis.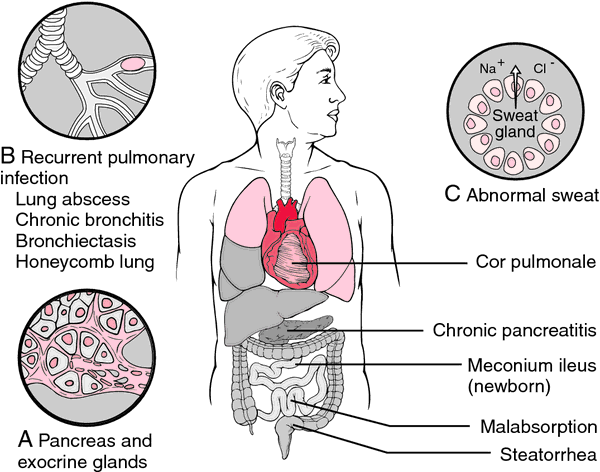
Effects. The symptoms and severity vary widely. Although cystic fibrosis is congenital, it may not manifest itself significantly during the early weeks or months of life, or it may cause intestinal obstruction and perforation in the newborn. The chief cause of complications is the extremely thick mucus produced. Normal mucus bathes and protects internal surfaces, transports chemicals produced in one organ through intricate small ducts to another organ, and carries bacteria, dirt, and wastes to be eliminated from the body; thus it needs to flow easily. The mucus of cystic fibrosis, in contrast, is highly adhesive. Bacteria and other matter stick to it, and it in turn clogs the lungs and usually interferes with the flow of digestive enzymes from the pancreas to the small intestine.
In the lungs, the mucus blocks the bronchioles, creating breathing difficulties. Infection develops, thereby increasing obstruction of the air passages. Air becomes trapped in the lungs (emphysema), and scattered small areas eventually collapse (patchy atelectasis). Repeated infections follow, inflaming and damaging lung tissue and leading to chronic lung disease. The organism that produces infection in cystic fibrosis is almost always a staphylococcus, but other organisms may be present in more severe cases. About half of children with cystic fibrosis have lung-related symptoms.
When mucus prevents the pancreatic enzymes from reaching the duodenum (as in about 80 per cent of cystic fibrosis patients), digestion is hindered. Fats especially are poorly digested and absorbed. The child may have a voracious appetite, yet fail to grow normally or gain weight. There may be marked signs of malnutrition. The outstanding symptom associated with pancreatic enzyme deficiency is frequent bulky, fatty, and foul-smelling feces.
Between 5 and 10 per cent of cystic fibrosis babies are born with intestines obstructed by puttylike intestinal secretions (meconium ileus) and die unless the condition is diagnosed promptly and relieved by surgery within the first few days of life. Such relief does not protect the child against the other manifestations of cystic fibrosis, although these may not appear until later.
Because cysts and scar tissue on the pancreas were observed during autopsy when the disease was first being differentiated from other conditions, it was given the name cystic fibrosis of the pancreas. Although this term describes a secondary rather than primary characteristic, it has been retained.
In the lungs, the mucus blocks the bronchioles, creating breathing difficulties. Infection develops, thereby increasing obstruction of the air passages. Air becomes trapped in the lungs (emphysema), and scattered small areas eventually collapse (patchy atelectasis). Repeated infections follow, inflaming and damaging lung tissue and leading to chronic lung disease. The organism that produces infection in cystic fibrosis is almost always a staphylococcus, but other organisms may be present in more severe cases. About half of children with cystic fibrosis have lung-related symptoms.
When mucus prevents the pancreatic enzymes from reaching the duodenum (as in about 80 per cent of cystic fibrosis patients), digestion is hindered. Fats especially are poorly digested and absorbed. The child may have a voracious appetite, yet fail to grow normally or gain weight. There may be marked signs of malnutrition. The outstanding symptom associated with pancreatic enzyme deficiency is frequent bulky, fatty, and foul-smelling feces.
Between 5 and 10 per cent of cystic fibrosis babies are born with intestines obstructed by puttylike intestinal secretions (meconium ileus) and die unless the condition is diagnosed promptly and relieved by surgery within the first few days of life. Such relief does not protect the child against the other manifestations of cystic fibrosis, although these may not appear until later.
Because cysts and scar tissue on the pancreas were observed during autopsy when the disease was first being differentiated from other conditions, it was given the name cystic fibrosis of the pancreas. Although this term describes a secondary rather than primary characteristic, it has been retained.
Diagnosis. Sweat in cystic fibrosis is excessively salty. Collapse of cystic fibrosis patients from salt loss during a heat wave led to recognition of the sweat abnormality. The sweat chloride test remains the cornerstone of diagnosis of cystic fibrosis. In children, a sweat chloride level above 60 mEq/l indicates cystic fibrosis. Newborn infants do not produce sufficient sweat for the test, but it has been noted that older babies “taste salty” when kissed. Supporting evidence can confirm the sweat test finding.
Treatment. Under careful supervision by the health care team, parents are taught the principles of home treatment of cystic fibrosis. The child may be required to sleep regularly in a plastic mist tent, into which a dense fog is pumped to help liquefy mucus and check infection. Aerosol therapy is generally prescribed. Extracts of animal pancreas taken with meals, which should be high in protein and low in fat, compensate for pancreatic deficiency. Physical therapy involving postural drainage together with “clapping” and “vibrating” aids in loosening the mucus so that it can be coughed up and expectorated.
It is estimated that in the United States cystic fibrosis occurs once in every few thousand births. Caucasians appear more subject to it than blacks, and among those of Asian descent it seems to be rare.
It is estimated that in the United States cystic fibrosis occurs once in every few thousand births. Caucasians appear more subject to it than blacks, and among those of Asian descent it seems to be rare.
Patient Care. Maintenance of the child's nutritional status may be difficult because of his tendency to cough and vomit frequently during feedings, and also because of difficulty in breathing. Small amounts of food, given slowly and at frequent intervals, are best for infants as well as for small children with cystic fibrosis.
Skin care is important, especially for infants and toddlers who are not yet toilet trained. The stools are likely to be copious and extremely irritating to the skin. Frequent turning of the infant or bedridden child helps to prevent decubitus ulcers and lessens the danger of pneumonia, a constant threat to these children. Prevention of infection is a most important aspect of the care of these children because of their extreme vulnerability to disorders of the respiratory tract.
Education of the parents must include the dietary regimen; how to use the Croupette, a machine for aerosol therapy in the home; hygienic measures to prevent infections; and the importance of continuous medical follow-up and administration of medications prescribed for the child.
Skin care is important, especially for infants and toddlers who are not yet toilet trained. The stools are likely to be copious and extremely irritating to the skin. Frequent turning of the infant or bedridden child helps to prevent decubitus ulcers and lessens the danger of pneumonia, a constant threat to these children. Prevention of infection is a most important aspect of the care of these children because of their extreme vulnerability to disorders of the respiratory tract.
Education of the parents must include the dietary regimen; how to use the Croupette, a machine for aerosol therapy in the home; hygienic measures to prevent infections; and the importance of continuous medical follow-up and administration of medications prescribed for the child.

Cystic fibrosis. The abnormal chloride transport associated with cystic fibrosis in a lack of sodium chloride in the secretions of all exocrine glands, especially the pancreas, intestine, and bronchi. Redrawn from Damjanov, 2000.
cystic kidney disease (cystic disease of kidney) see acquired cystic kidney disease and polycystic kidney disease.
fibrosis
[fi-bro´sis]formation of fibrous tissue; see also fibroid degeneration. adj., adj fibrot´ic.
congenital hepatic fibrosis a developmental disorder of the liver, marked by formation of irregular broad bands of fibrous tissue containing multiple cysts formed by disordered terminal bile ducts, resulting in vascular constriction and portal hypertension.
cystic fibrosis (cystic fibrosis of pancreas) see cystic fibrosis.
diffuse idiopathic interstitial fibrosis (diffuse interstitial pulmonary fibrosis) idiopathic pulmonary fibrosis.
endomyocardial fibrosis an idiopathic type of myocardiopathy that is endemic in various parts of Africa and rarely in other areas, characterized by cardiomegaly, marked thickening of the endocardium with dense white fibrous tissue that may extend to involve the inner myocardium, and by congestive heart failure.
idiopathic pulmonary fibrosis chronic inflammatory progressive fibrosis of the pulmonary alveolar walls, with steadily progressive dyspnea, resulting in death from oxygen lack or right heart failure. Most cases are of unknown origin, although some are thought to result from pneumoconiosis, hypersensitivity pneumonitis, scleroderma, and other diseases.
mediastinal fibrosis development of hard white fibrous tissue in the upper portion of the mediastinum, sometimes obstructing the air passages and large blood vessels; called also fibrosing or fibrous mediastinitis.
periureteral fibrosis retroperitoneal fibrosis.
pleural fibrosis fibrosis of the visceral pleura so that part or all of a lung becomes covered with a plaque or a thick layer of nonexpansible fibrous tissue. The more extensive form is called fibrothorax.
postfibrinous fibrosis that occurring in tissues in which fibrin has been deposited.
proliferative fibrosis that in which the fibrous elements continue to proliferate after the original causative factor has ceased to operate.
pulmonary fibrosis idiopathic pulmonary fibrosis.
retroperitoneal fibrosis deposition of fibrous tissue in the retroperitoneal space, producing vague abdominal discomfort, and often causing blockage of the ureters, with resultant hydronephrosis and impaired renal function, which may result in renal failure. Called also Ormond disease.
fibrosis u´teri a morbid condition characterized by overgrowth of the smooth muscle and increase in the collagenous fibrous tissue of the uterus, producing a thickened, coarse, tough myometrium.
Miller-Keane Encyclopedia and Dictionary of Medicine, Nursing, and Allied Health, Seventh Edition. © 2003 by Saunders, an imprint of Elsevier, Inc. All rights reserved.
cys·tic fi·bro·sis
, cystic fibrosis of the pancreas [MIM*219700]a congenital metabolic disorder in which secretions of exocrine glands are abnormal; excessively viscid mucus causes obstruction of passageways (including pancreatic and bile ducts, intestines, and bronchi), and the sodium and chloride content of sweat is increased throughout the patient's life; symptoms usually appear in childhood and include meconium ileus, poor growth despite good appetite, malabsorption and foul bulky stools, chronic bronchitis with cough, recurrent pneumonia, bronchiectasis, emphysema, clubbing of the fingers, and salt depletion in hot weather. Detailed genetic mapping and molecular biology have been accomplished by the methods of reverse genetics; autosomal recessive inheritance, caused by mutation in the cystic fibrosis conductance regulator gene (CFTR) on chromosome 7q.
Synonym(s): Clarke-Hadfield syndrome, fibrocystic disease of the pancreas, mucoviscidosis, viscidosis
Farlex Partner Medical Dictionary © Farlex 2012
mucoviscidosis
(myo͞o′kō-vĭs′ĭ-dō′sĭs)n.
See cystic fibrosis.
The American Heritage® Medical Dictionary Copyright © 2007, 2004 by Houghton Mifflin Company. Published by Houghton Mifflin Company. All rights reserved.
mucoviscidosis
Cystic fibrosis, see there.McGraw-Hill Concise Dictionary of Modern Medicine. © 2002 by The McGraw-Hill Companies, Inc.
cys·tic fi·bro·sis
, cystic fibrosis of the pancreas (sis'tik fī-brō'sis, pan'krē-ăs) [MIM*219700]Congenital metabolic disorder in which secretions of exocrine glands are abnormal; excessively viscid mucus obstructs passageways; the sodium and chloride content of sweat is increased throughout the patient's life.
Medical Dictionary for the Dental Professions © Farlex 2012
Patient discussion about mucoviscidosis
Q. Can cystic fibrosis patients have children? My boyfriend has cystic fibrosis, and currently he’s treated with many medications but usually healthy (other than pneumonia from hospitalization from time to time). I heard that men with cystic fibrosis can’t have children - is that true? Is there anything he can do about it?
A. WE ARE A HEALTHY COUPLE BUT MY WIFE NEVER GET PREGNANT IN 20 YEARS OF RELATIONSHIP.
I'M SO SORRY ABOUT MY ENGLISH, I'M LEARNING.
I'M SO SORRY ABOUT MY ENGLISH, I'M LEARNING.
Q. Do women with cystic fibrosis have difficult pregnancy? My wife has cystic fibrosis, and after 3 year of marriage we decided we want a baby. I know that men with cystic fibrosis are usually infertile and can’t have children- is that the case also for women with cystic fibrosis? Is the pregnancy in women with cystic fibrosis more problematic? Is it dangerous?
A. Before you attempt a pregnancy, you should consult her doctor to make sure she can tolerate it, because very severe disease can make the pregnancy dangerous for her. If her disease isn’t so severe, usually there are no special problems.
More discussions about mucoviscidosisThis content is provided by iMedix and is subject to iMedix Terms. The Questions and Answers are not endorsed or recommended and are made available by patients, not doctors.
Latest Searches:
zygomaticofrontal - zygomaticofacialis - zygomaticofacial - zygomaticoauricular - zygomatico - zygomatici - zygomatica - zygomatic - zygoma - zygomas - zygodactyly - Zygocotyle - zygion - zygia - zygapophysis - zygapophysiales - zygapophysial - zygapophyseales - zygapophyseal - zygal -
- Service manuals - MBI Corp